Viewpoint - Annals of Biological Research ( 2018) Volume 9, Issue 1
Nickel is a ubiquitous environmental transition metal that is widely used in industry. Epidemiological studies indicate that chronic occupational exposure to nickel (Ni) compounds induces human lung and nasal cancers [1]. So far,however, little is known about molecular mechanisms of nickel-induced malignant transformation and tumor development. This study shows that the Nrf2 (nuclear factor erythroid 2-related factor 2) is highly expressed in both human lung tumor tissue and in nickel-transformed human lung bronchial epithelial BEAS-2B cells. Basal ROS(reactive oxygen species) levels are extremely low in nickel-transformed BEAS-2B cells and this correlates with elevated expressions of both antioxidant enzymes and anti-apoptotic proteins. Low levels of ROS and expression levels of antioxidant enzymes and anti-apoptotic proteins are tightly regulated by Nrf2. Autophagy inhibition,induced by pharmacologically or genetically, enhanced nickel-induced apoptosis, indicating that the induction of autophagy is the cause of apoptosis resistance in nickel-transformed cells. This study also shows that in nickel-transformed cells, inhibition of apoptosis cause decreases autophagy in nickel-transformed cells. Additionally, Stat3,which is up-regulated by Nrf2, controls autophagy induction in nickel-transformed cells. Collectively, this study demonstrates that constitutive high Nrf2 expression parallel inhibits apoptosis by an up-regulation of antioxidant enzymes and anti-apoptotic proteins, and increases autophagy via Stat3 signalling
Nrf2, Apoptosis, Autophagy, Nickel, Transformed cells
Nrf2: Nuclear Factor Erythroid 2-Related Factor 2; ROS: Reactive Oxygen Species; bzip: Basic Leucine Zipper; IARC: International Agency for Research on Cancer; SOD: Superoxide Dismutase; ChIP: Chromatin Immunoprecipitation
Nickel is a ubiquitous environmental transition metal that is widely used in industry. Epidemiological studies indicate that chronic occupational exposure to nickel (Ni) compounds induces human lung and nasal cancers. So far, however, little is known about molecular mechanisms of nickel-induced malignant transformation and tumor development. This study shows that the Nrf2 (nuclear factor erythroid 2-related factor 2) is highly expressed in both human lung tumor tissue and in nickel-transformed human lung bronchial epithelial BEAS-2B cells. Basal ROS (reactive oxygen species) levels are extremely low in nickel-transformed BEAS-2B cells and this correlates with elevated expressions of both antioxidant enzymes and anti-apoptotic proteins. Low levels of ROS and expression levels of antioxidant enzymes and anti-apoptotic proteins are tightly regulated by Nrf2. Autophagy inhibition, induced by pharmacologically or genetically, enhanced nickel-induced apoptosis, indicating that the induction of autophagy is the cause of apoptosis resistance in nickel-transformed cells. This study also shows that in nickel-transformed cells, inhibition of apoptosis cause decreases autophagy in nickel-transformed cells. Additionally, Stat3, which is upregulated by Nrf2, controls autophagy induction in nickel-transformed cells. Collectively, this study demonstrates that constitutive high Nrf2 expression parallel inhibits apoptosis by an up-regulation of antioxidant enzymes and antiapoptotic proteins, and increases autophagy via Stat3 signalling [1].
Industrial or environmental exposure to nickel compounds is associated with a higher incidence of human lung cancer. As a result, nickel compounds were classified as human carcinogens by the International Agency for Research on Cancer (IARC) in 1990 [2]. Nickel has weak mutagenic effects and could transform cells by acting on a number of target molecules. However, the precise mechanisms underlying its carcinogenic activity are not fully understood.
Nrf2 is a basic leucine zipper (bZIP) transcription factor that serves as a master regulator of cellular redox homeostasis [3,4]. Nrf2 binds to antioxidant response elements in gene promoters and transcriptionally activates antioxidant enzymes as well as some apoptosis regulatory proteins. Constitutive activation of Nrf2 contributes to malignant transformation, and high expression of Nrf2 has been observed in a variety of tumor cells as well as cadmium- or arsenic-induced transformed cells [5,6]. This study shows that Nrf2 expression was markedly higher in lung tumor tissues from patients with a 40-year smoking history. While BEAS-2B human lung bronchial epithelial cells that had been chronically exposed to nickel produced tumors, BEAS-2B cells in which Nrf2 expression had been suppressed by shRNA against Nrf2 prior to the nickel exposure produced smaller tumors in mice xenografts model. Silencing Nrf2 expression by either siRNA or shRNA against Nrf2 also impaired colony formation by BEAS-2B cells that had been chronically exposed to nickel. Collectively, these results support the pathological relevance of Nrf2 signaling in nickel-induced carcinogenesis. The generated nickel- transformed cell by continuously exposing BEAS-2B cells to 50 μM Ni2+ for four months, Nrf2 expression was higher when compared with the normal BEAS-2B cells. Furthermore, in nickel-transformed cells, the expression of Nrf2 was accompanied with resistance to apoptosis. Nickel-transformed cells also had a higher proliferating potential than their parental cell line. Interestingly, nickel-transformed cells have lower ROS levels and a higher level of the antioxidant enzymes catalase, SOD1 (superoxide dismutase1) and SOD2. These low levels of ROS and high expressions of antioxidant enzymes are involved in the resistance of nickel-transformed cells to apoptosis. Since siRNA-mediated knockdown of Nrf2 in nickel-transformed cells enhanced the generation of ROS and attenuated the expression and activities of catalase, SOD1 and SOD2, those results conclude that Nrf2 tightly regulates antioxidant enzymes and contributes to maintaining low a level of ROS in these cells. Furthermore, compared with BEAS-2B cells, nickel-transformed cells express high levels of the antiapoptotic proteins Bcl-2 and Bcl-xL, contributing to the resistance of nickeltransformed cells to apoptosis. ChIP (Chromatin immunoprecipitation) analysis revealed that Nrf2 binds to the ARE (antioxidant response element) containing regions of the Bcl-2 or Bcl-xL promoter and regulates their transcription. Moreover, siRNA-mediated knockdown of Nrf2 expression in nickel-transformed cells reduced the expressions of Bcl-2 and Bcl-xL. This suggests the up-regulation of anti-apoptotic proteins by Nrf2 might play a major role in the apoptosis resistance of nickel-transformed cells. Our findings suggest that, by enhancing the levels of anti-apoptotic proteins, Nrf2 acts as a survival factor in the nickel-transformed cells.
This study also found that treatment of nickel increased the levels of LC3-II in nickel-transformed cells and to a greater extent than in BEAS-2B cells, indicating that nickel transformed are sensitive to autophagy induction following nickel exposure. This phenomenon might help nickel transformed to resist apoptosis. This result was further confirmed by several kinds of autophagic flux assays. Nickel-induced cell viability was attenuated, and apoptosis was enhanced, by inhibiting autophagy using both pharmacological and genetic approaches in nickeltransformed cells. This study found five consensuses AREs in the Stat3 promoter, among ARE-containing regions of the Stat3 promoter, Nrf2 able to binds two ARE binding regions. The extent of Nrf2 binding of these regions was higher in nickel-transformed cells than in non-malignant BEAS-2B cells. Furthermore, siRNA-mediated knockdown of Nrf2 in nickel-transformed cells completely depleted the levels of phosphorylated Stat3 and attenuated the total Stat3 levels. These results indicate that the high basal level of Stat3 in NiT cells directly results from Nrf2-mediated transcriptional activation. These results assume that the Nrf2-induced up-regulation of Stat3 contributes to the autophagy sensitivity and apoptosis resistance. In BEAS-2B cells, overexpressing of Stat3 increased LC3-II levels as well as the number of cells containing GFP-LC3 puncta, while inhibition of Stat3 completely abolished the nickelinduced increase in LC3-II levels and in the number of cells containing GFP-LC3 puncta in nickel-transformed cells. This study confirmed that autophagy contributes to cell survival in nickel-transformed cells. These results suggest that autophagy and apoptosis have a negative feedback loop in cancer cells and that the regulations of these mechanisms are important for chemotherapeutic strategies.
In conclusions, nickel-transformed cells have properties of low ROS levels and apoptosis resistance by up-regulating antioxidant enzymes and anti-apoptotic proteins. The sensitivity of nickel-transformed cells to autophagy induction enhances their resistance to apoptosis. In nickel transformed cell, high expression of Stat3 is responsible for the increased autophagy. Antioxidant enzymes, anti-apoptotic proteins, and Stat3 are tightly regulated by Nrf2 in the transformed cells. Also, Nrf2 is highly expressed in human lung tumor tissues as well as nickel-transformed cells. Collectively, our results reveal that tumor or transformed cell survival mechanisms involve the down-regulation of apoptosis and up-regulation of autophagy. Furthermore, Nrf2 is a coordinate regulator of intracellular ROS levels, apoptosis resistance, autophagy sensitivity in nickel-transformed cells; these phenomena contribute to cell survival and carcinogenesis (Figure 1). Nrf2 appears to be a double-edged sword having both tumor-suppressing and tumorpromoting functions. The model of the nickel-transformed cell well describes these paradoxical effects of Nrf2 and provides two anticancer therapeutic approaches. The first prevention strategy is to increase the Nrf2 for inhibiting ROS generation in normal cells. The opposite approach of anticancer therapy is inhibiting Nrf2 expression for promoting ROS generation in tumor cells. Nrf2-inducing for normal tissue and Nrf2-inhibiting for a tumor can be effective strategies for both tumor prevention and therapy.
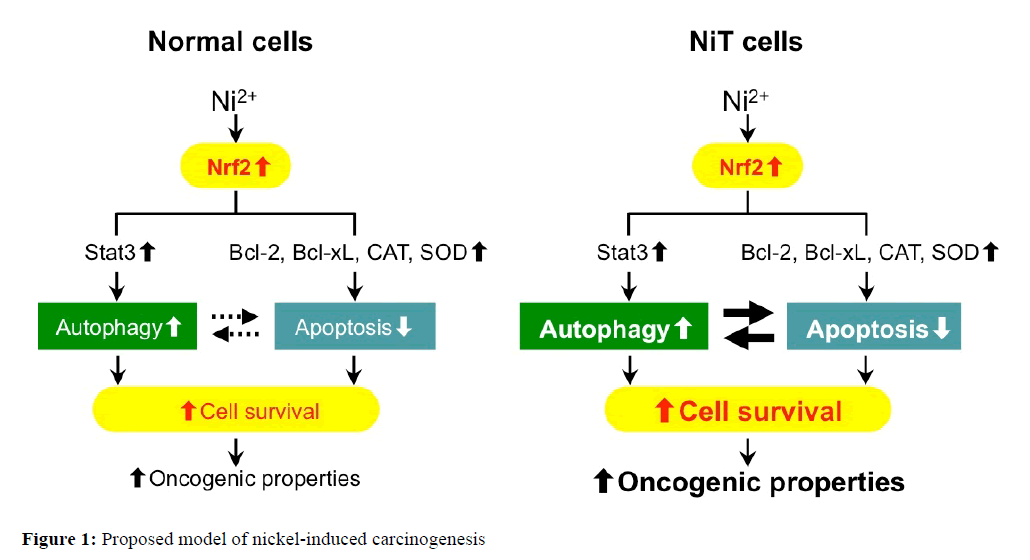
Figure 1: Proposed model of nickel-induced carcinogenesis
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (2016R1D1A1B03930327).